The NOE module uses the full relaxation matrix method (CORMA) to calculate NOE intensities from internuclear distances. This tutorial will show you how the NOE module works with the quinine molecule:
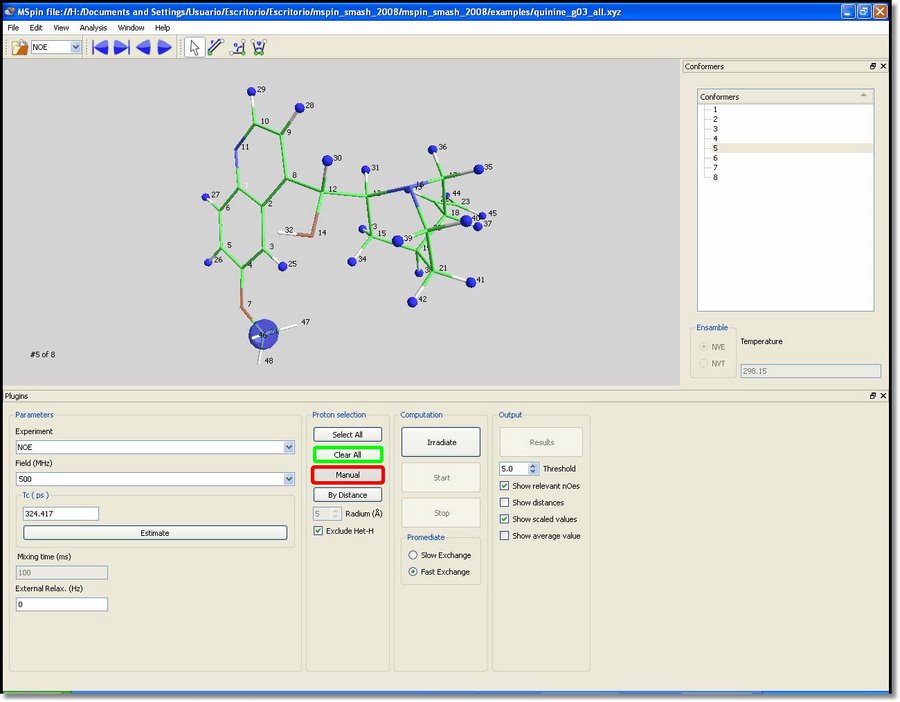
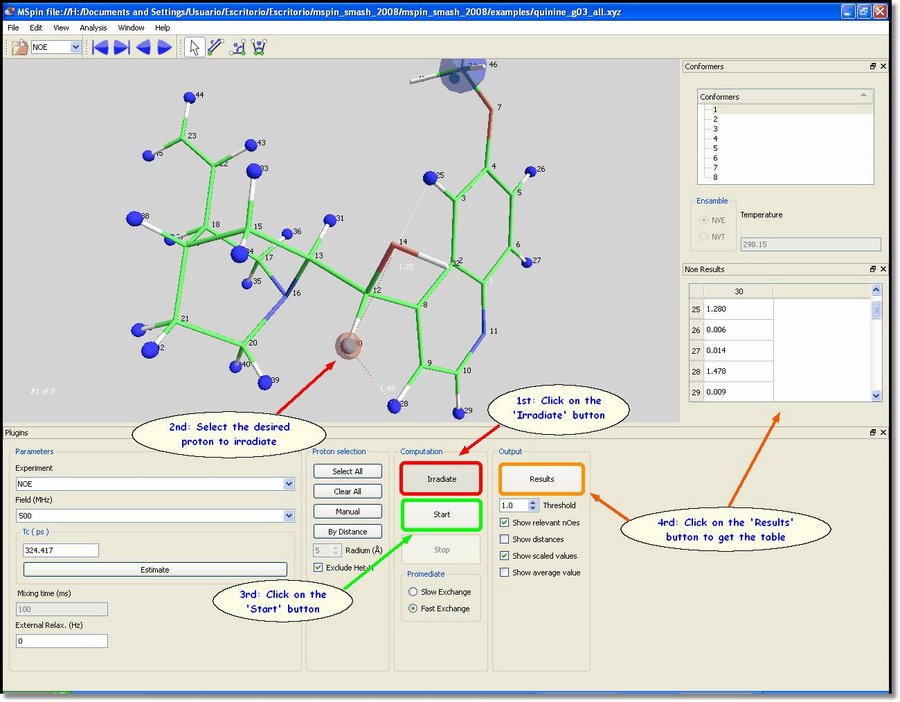
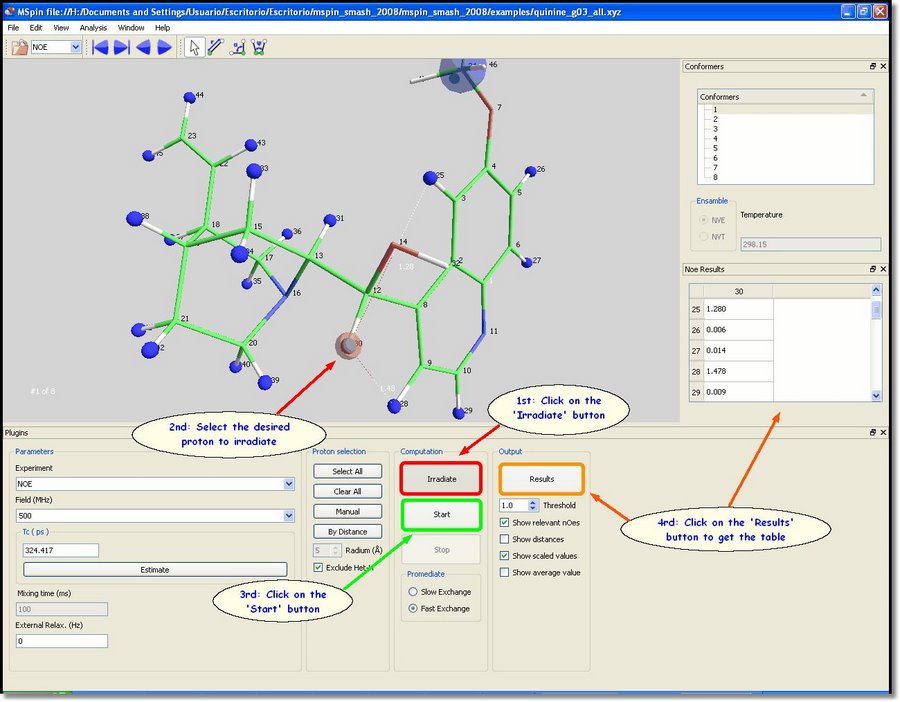
All hydrogens (except the interchangeable ones, such as hydroxyl groups) will be highlighted in blue when the user loads a molecular structure in the NOE module. These blue atoms will be included in the NOE calculation. The methyl groups will be also considered as single particles during computation, so they will also be highlighted in blue. The user will be able to deselect all atoms just by clicking on the 'Clear All' button (green rectangle in the picture above). Alternatively, you can select/deselect atoms one by one by clicking on each proton after having clicked on the '˜Manual' button (red rectangle in the picture below). We are going to select only the proton number 30. With all the atoms of the molecule highlighted in blue, click on the '˜Irradiate' button and then select the proton number 30 (which will be highlighted in red). Finally click on the 'Start' button to perform the computation and then on the 'Results' button to show the results in a table, as you can see in the picture below:
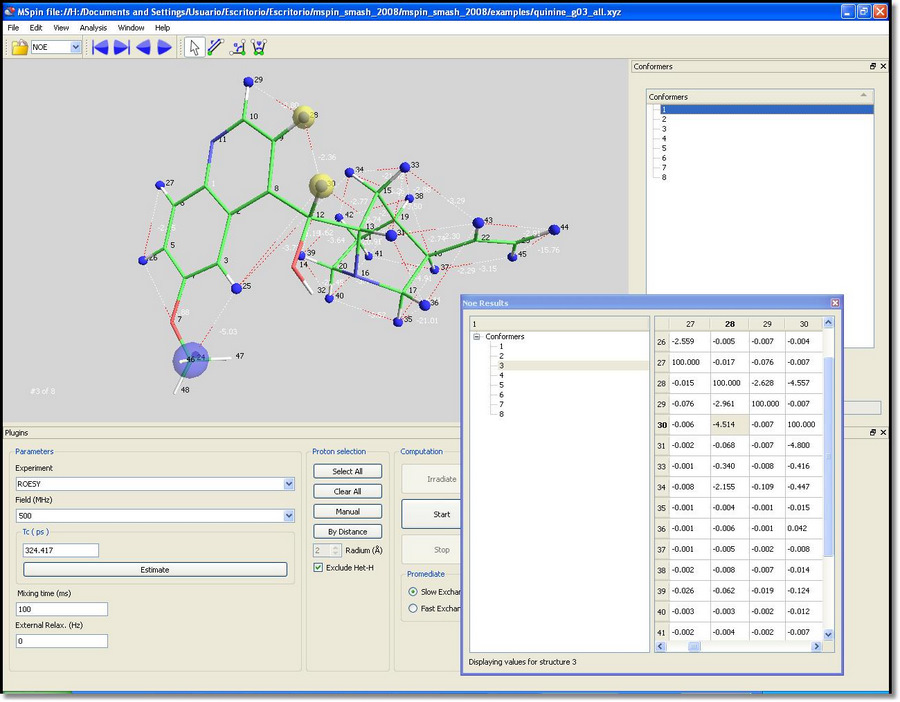
You can click on the cells of the table to highlight the atoms involved in the NOE:
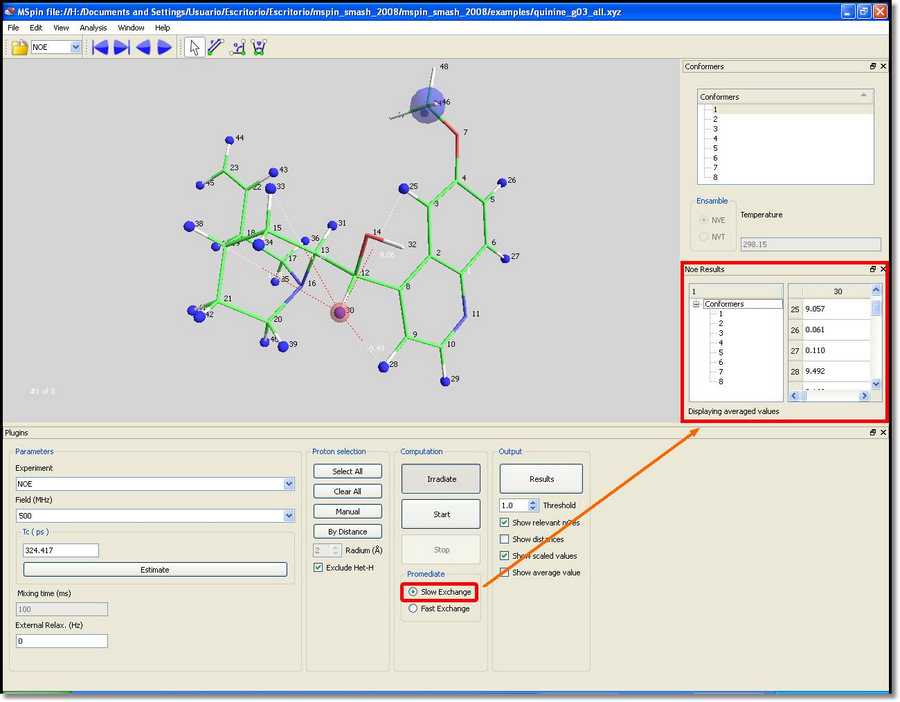
You can also select the 'low exchange option' and see how individual values are computed for each conformer:
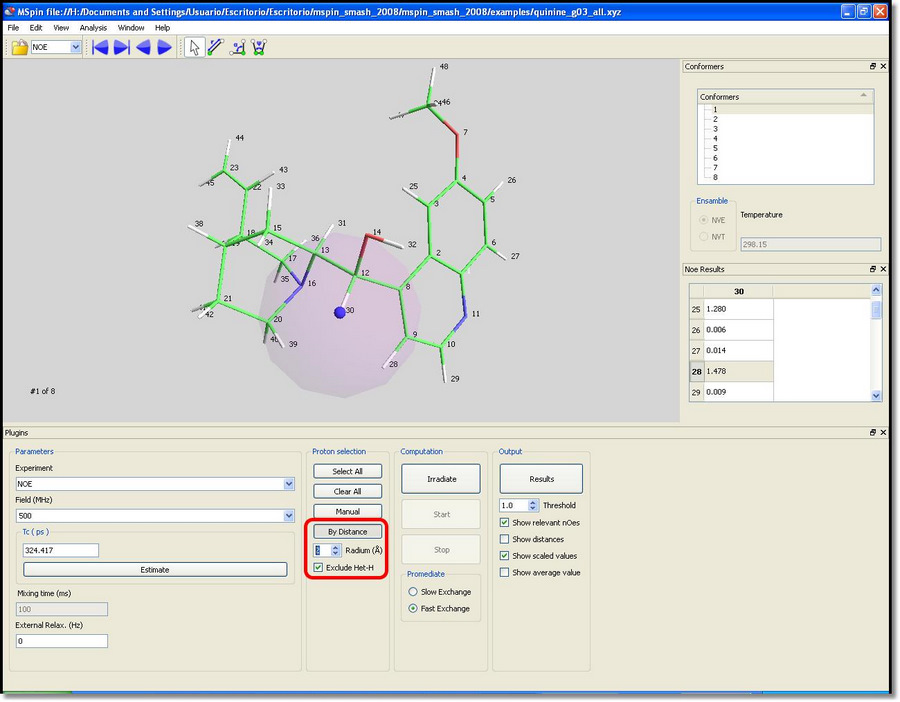
Frequently, the user may want to perform computation only on a region of interest in a large molecule. This can be done using the 'By Distance' button. Clicking on this button and picking on a proton, all atoms inside a sphere (shown in purple) of the desired radium, will be included in the computation.
Once the desired spin system has been selected, the user will also be able to select the type of experiment in the corresponding scroll down menu (Stationary NOE, NOESY or ROESY experiments), the Field (in MHz), the correlation time (by default the molecular mass in picoseconds), the mixing time for NOESY and ROESY experiments and the external relaxation velocity ??, which can be adjusted to fit the experimental to the theoretical results. At this point, the user will be able to proceed through the computation. In the case of a NOESY- ROESY computation, just click on the 'Start' button. By default, all cross-peaks above the selected threshold will be shown on screen. Additionally, the intermolecular proton-proton distances can be also shown on screen. A table with the whole intensities matrix can be shown by clicking on the 'Result' button. Note that clicking on any cell will display a cross-line between the two spins related to that cross-term. From this window results can be printed or exported to the clipboard just by right clicking. In this case, we have selected a ROESY experiment with 'low exchange':