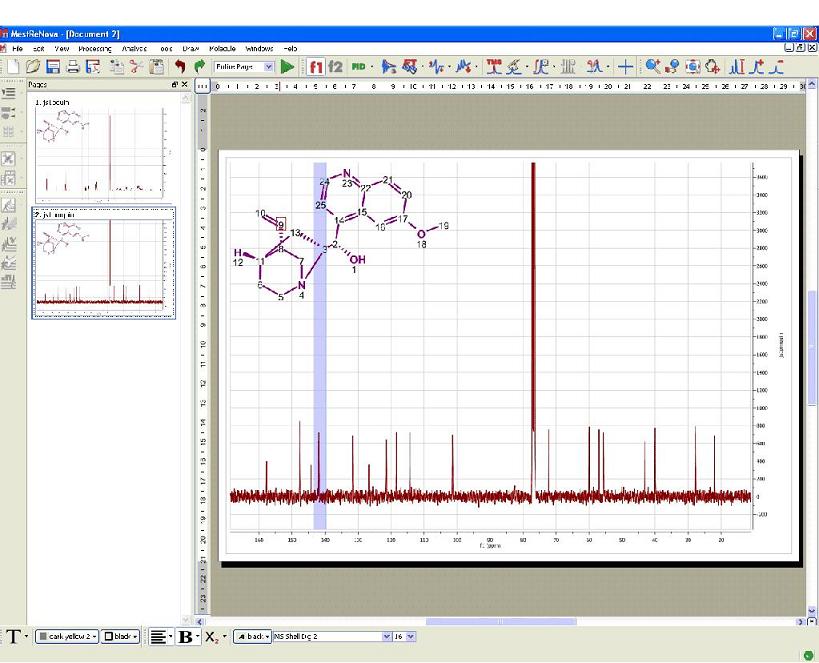
This feature will calculate in the background a simulation of the spectrum of the molecular structure present in the spectral window, highlighting the expected chemical shifts when the user hovers the mouse over a proton or a carbon. This tool will be very useful to help the user in the process of assigning 1D NMR spectra. The user will be able to use this tool with 1H and 13C NMR spectra just by pasting the corresponding molecular structure (quinine, in this example) over the spectrum and following the menu 'Analysis/Predict & Highlight/Predict ', as shown in the picture below:

The algorithm run for the simulation carried out in the background will be the one selected in the 'Molecule/Prediction Options' menu. The user will be able to select the 'Increments' or the 'Charge' algorithm in the 1H simulation and the 'Neural Network' system (by using NMRPredict Desktop) or the HOSE database methodology (implemented in the server-based NMR Predict application only). After having applied this feature, the user will notice that hovering the mouse over an atom will highlight the area on the spectrum corresponding to the simulated value for that atom. The user will therefore get an indication as to the fact that experimental signals falling within the highlighted area may correspond with the proton directly bonded to the carbon number 9 on the molecule.
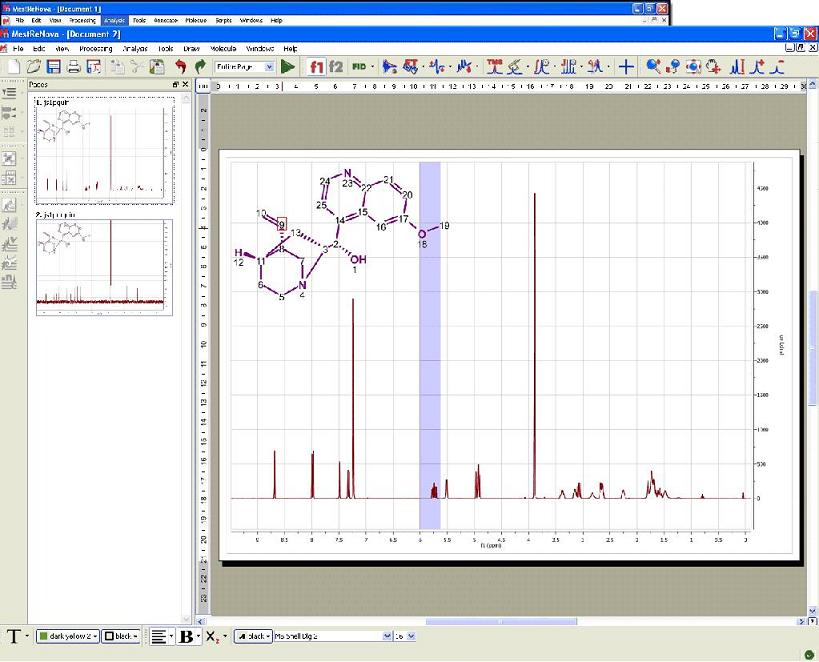
The user will be able to apply the same feature with the 13C NMR spectra, as shown in the picture below: