

Mnova 11 is a major release that incorporates many new features in NMR, MS, Molecule Editor, Database, Verify and NMRPredict plugins. We have integrated a new PhysChem properties module as well as important enhancements in our Scripting Engine. This version has fixed several bugs.
New & highly improved features
- 1D and 2D assignments have been considerably improved:
- Assisted Assignments.
- Correlation arrows in the molecule.
- Improved workflow.
- “Quality” of each assignment in the assignments table.
- ‘Best NMR’ Prediction.
- PhysChem properties module.
- Improved support of NUS experiments.
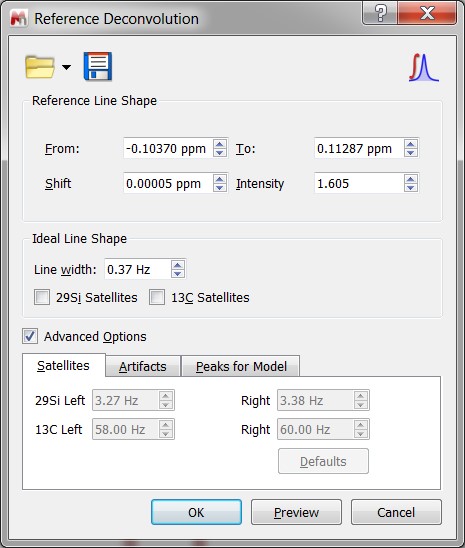
- Advanced Reference Deconvolution toolkit.
- DOSY processing:
- New DOSY processing algorithm.
- Non-Uniform Gradient (NUG) correction.
- Auto track moving peaks in Reaction Monitoring.
- Support of Covariance and Pure Shift Experiments for Advanced NMR processing.
- Popup molecule widget.
- Mnova MS peak purity calculation.
- New chemometrics module.
- NMR Assignements
-
- Assisted Assignments. After entering the Manual Assisted Assignment mode, if you hover the mouse over the atoms of the molecule, the suggested multiplets in the spectrum get highlighted. When hovering the mouse over the multiplets, the suggested atoms in Compounds accordionle get also highlighted.

- Capability to show 1H and 13C assignments in atom labels.

- Assignments transferred to multiplets when copying an assigned molecule to a new dataset.
- Do not remove all correlations involved when deleting one of the shifts in case of ambiguous assignments.
- Remove all correlations involved when deleting an assigned multiplet, peak or integral.
- Remove all correlations involved when deleting the shift value of an assignment.
- H-H correlations between diastereotopic protons are auto deduced.
- 2D correlations auto deductions are checked again after replacing or swaping assignments.
- Remove all correlations involved when deleting an assigned multiplet, peak or integral.
- When assigning symmetric Hs all the attached Cs get auto deduced from the HSQC correlation.
- Second H of a diastereotopic CH2 auto deduced from the HSQC.
- Ability to add overlapping multiplets by assigning to a 2D range.
- Many pop up messages removed during manual assignment workflow.
- Improved auto deduction routines to get derived assignments from 2D correlations.
- Manual assignment propagation from 13C dimension of 1H-13C correlation transferred to 1H spectrum.
- Manual assignment propagation from 1H to 13C dimension now happens when 1H assignment is made on a 2D 1H-1H correlation spectrum.
- Improved transfer of manual assignment from 1H to 13C dimension when cross peaks are close on the 1H dimension of the 2D spectrum.
- Help messages in the status bar when using Assignment tool.
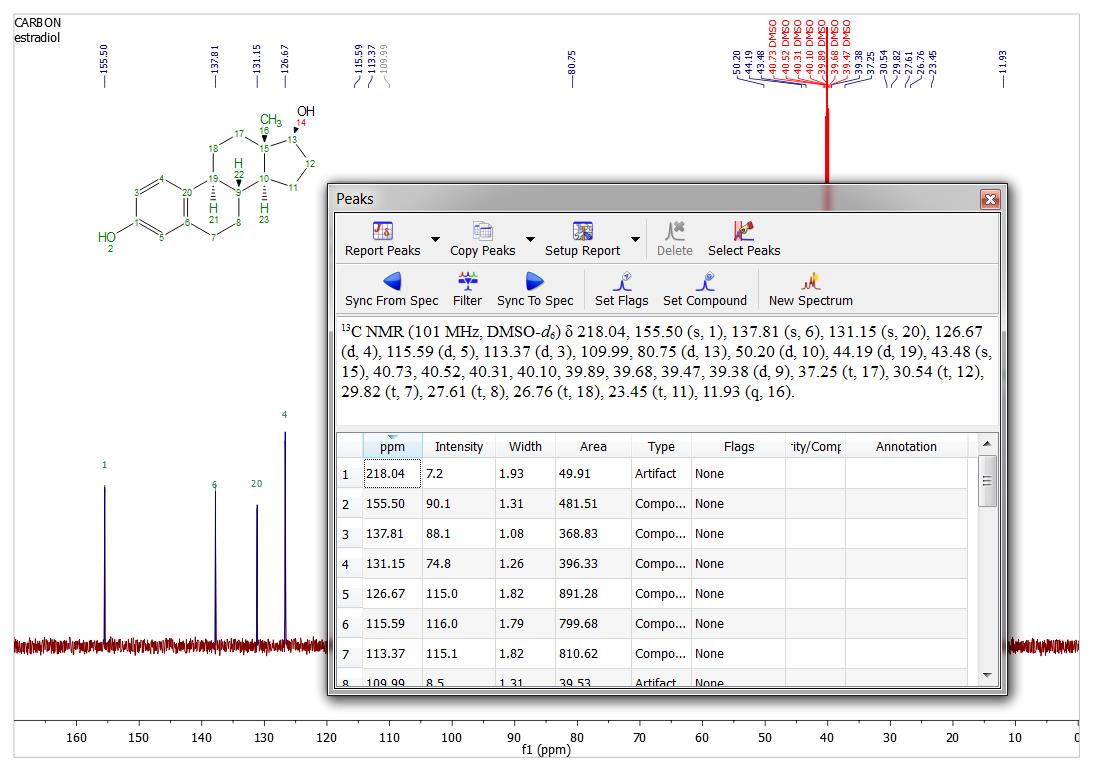
- Improvements in the report assignments script.
- Reports of assignments now switch to multiple columns when they are too long.
- Automatic recalculation of the 2D deductions after making some changes in the 2D spectra.
- In CH2 assignment if Ha is assigned show Hb label by default when assigning the second H.
- Ability to delete assignments and correlations graphically from the spectrum.
- Report 13C peaks with multiplicity. After making assignments to the 13C peaks the multiplicity gets automatically filled in Peaks accordionle. This multiplicity information can be included in the Peaks Report by selecting it in Setup Report.
- Assisted Assignments. After entering the Manual Assisted Assignment mode, if you hover the mouse over the atoms of the molecule, the suggested multiplets in the spectrum get highlighted. When hovering the mouse over the multiplets, the suggested atoms in Compounds accordionle get also highlighted.
- NMR Predict
-
- “Best NMR” prediction has been implemented to enhance our NMRPredict module.

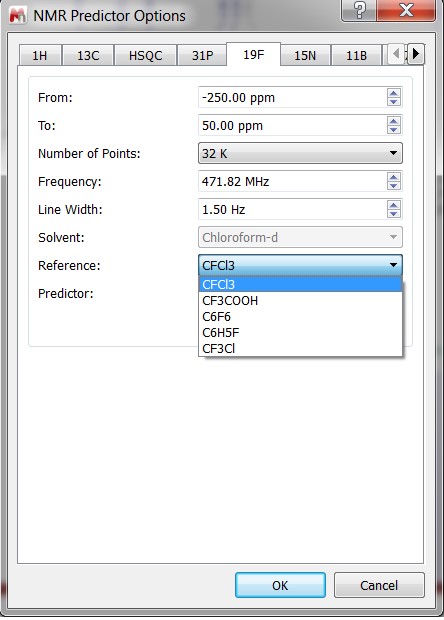
- Capability to select the default solvent reference in 19F and 15N NMR predictions.
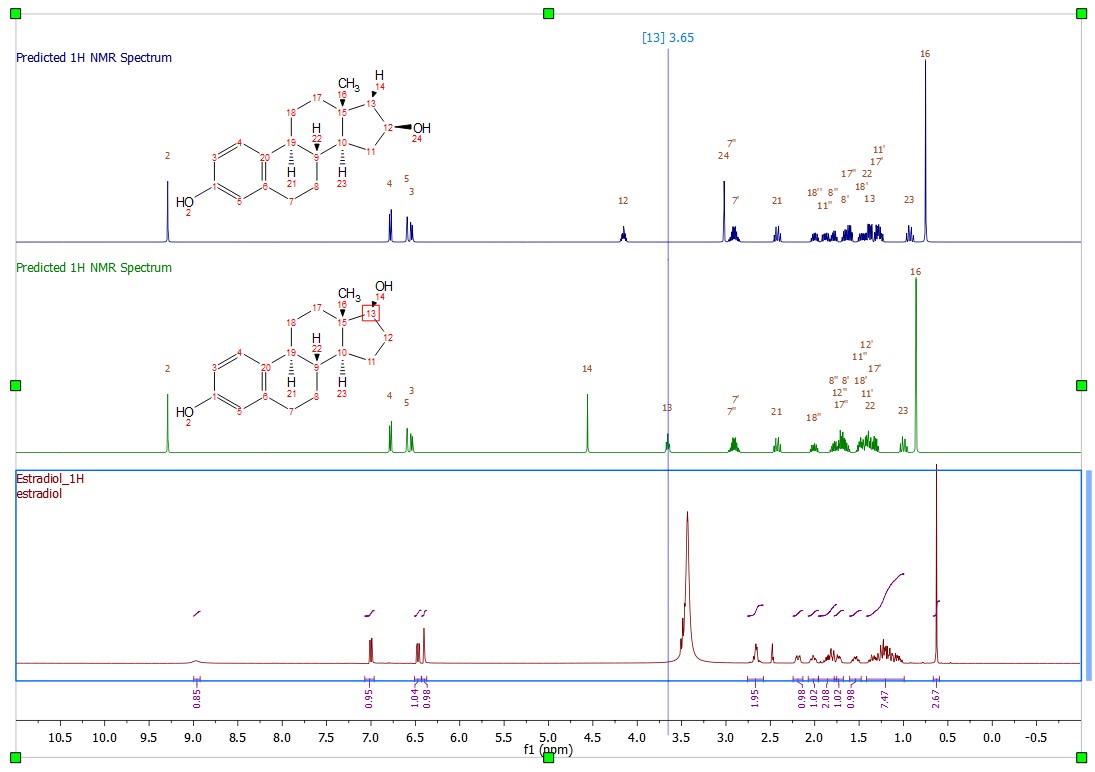
- Capability to update predicted spectra in stacked plots. Predicted spectra can be modified either graphically or by editing the values in Prediction accordionle or Spin Simulation panel. Both spectra and accordionles get correctly updated after switching between the predicted spectra. In case of multiple predicted spectra in the stacked, the data displayed in the Prediction accordionle and the Spin Simulation panel correspond to the active predicted spectrum.

- Ability to get highlighting like in Predict &Highlight using predictions read from a file via script.
- Capability to calculate 3J HNCH couplings.
- “Best NMR” prediction has been implemented to enhance our NMRPredict module.
- MS
-
- Capability to load/save the Peak Detection options.

- Capability to specify the desired resolution to build the centroid spectrum. The user can manually adjust the peak detection settings, allowing to specify the desired resolution to build the centroid spectrum.

- Capability to load/save the Elemental Composition and Molecular Match settings.


- Align traces automatically depending on the LCMS machine. This feature is useful to align UV/DAD and MS traces by using a correction value.
 Those parameters can be predefined from the ‘Edit/Preferences/Mass/Import’ menu:
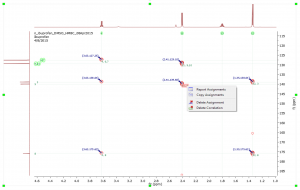
Those parameters can be predefined from the ‘Edit/Preferences/Mass/Import’ menu:  The trace alignment parameters are automatically applied to any new trace or DAD chromatogram and the default value for those parameters is determined by the selected preferences. When importing a dataset, if any of the rules matches, then the parameters present in the matched rule are copied to the new item. For the rules, you can specify to which provider (or any provider) the rule applies, and a set of parameter rules based on pattern matching:
The trace alignment parameters are automatically applied to any new trace or DAD chromatogram and the default value for those parameters is determined by the selected preferences. When importing a dataset, if any of the rules matches, then the parameters present in the matched rule are copied to the new item. For the rules, you can specify to which provider (or any provider) the rule applies, and a set of parameter rules based on pattern matching: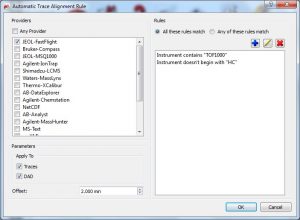
- Peak purity calculation. This feature will calculate the chromatogram peak (or region) purity associated to the most abundant mass peaks which are displayed under the selected peak: Ms peak purity.

- Improved vertical axis label for chromatograms. A Default checkbox has been added. When it’s checked, it means that the label will be set by Mnova to the default value for that kind of plot (so the label line editor remains disabled). If it’s unchecked, then the label will be set to whatever the user sets in the line editor.

- New converter Advion MS data.
- Capability to load/save the Peak Detection options.
- NMR
-
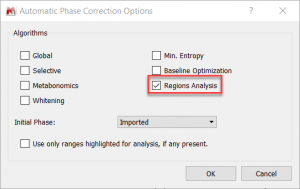
- 1D NMR autophase routine: New “Region analysis” algorithm. This new algorithm improves considerably the automatic phase routine for 1D NMR spectra. It can be selected on the Automatic Phase Correction options.
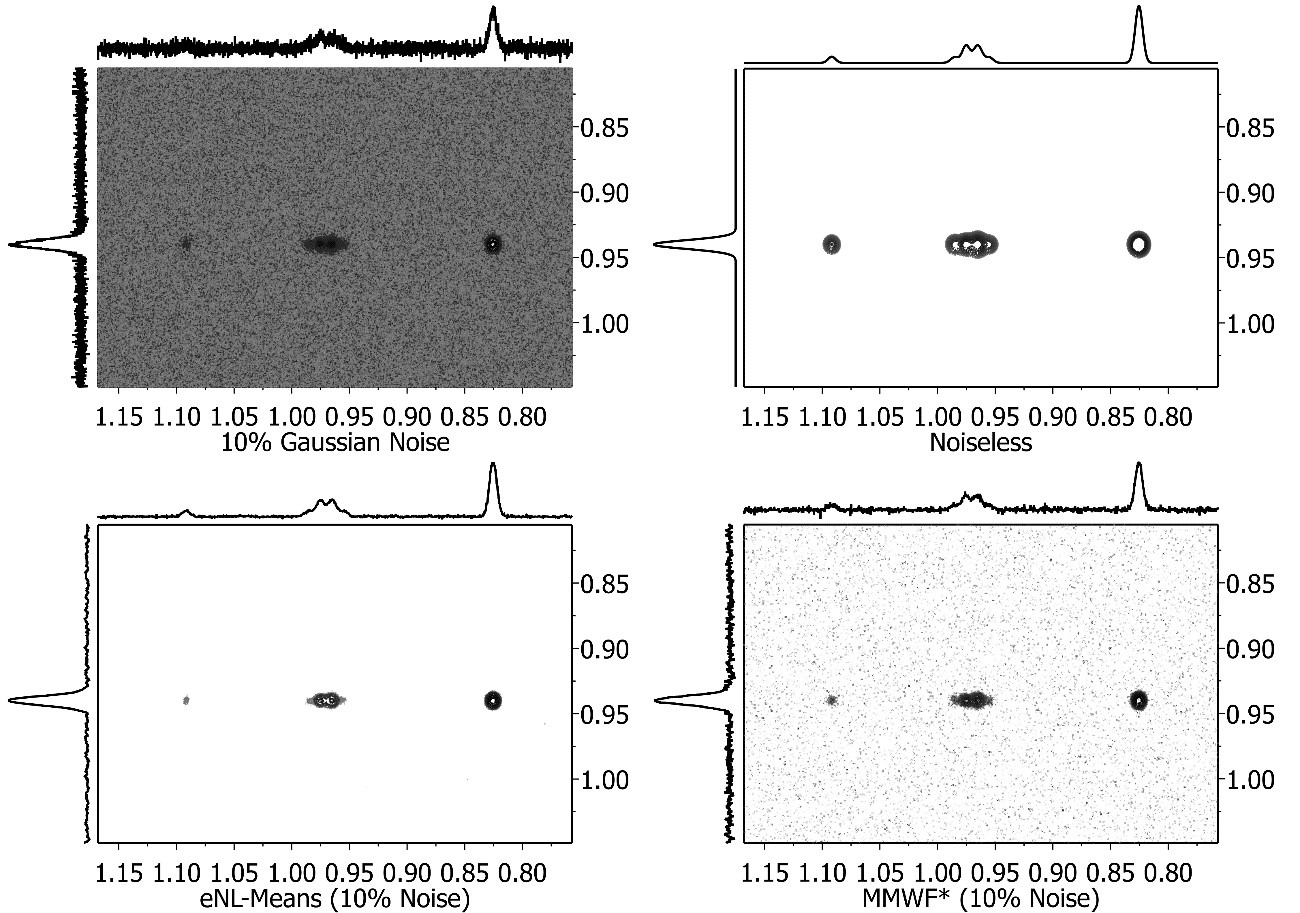
- Denoising: In any experimental technique, the distinction between signals and noise is a real factor, and this is particularly the case with NMR because of its low inherent sensitivity. The application of the so-called ‘spatial filters’ has been proposed for the efficient denoising of images as well as for 2D NMR data sets. Mnova includes a number of those filters aimed at reducing the noise level while maintaining the original spectral resolution.

- Use GSD knowledge in multipoint baseline correction.

- Improved Reference Deconvolution.
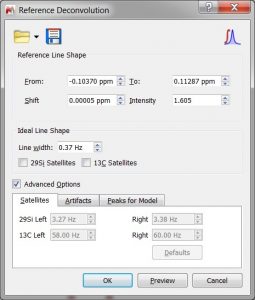
- Improved Group Delay Correction: Now this method can properly handle phase values imported from the spectrometer.
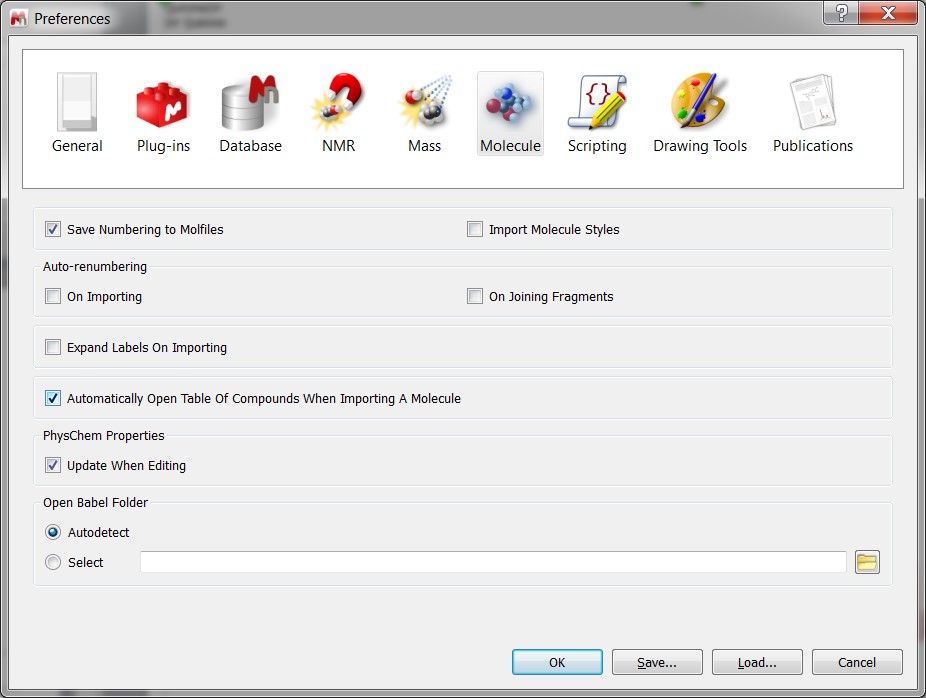
- New preference to pop-up the accordionle when opening a new molecule.

- Ability to apply layout templates to specific document pages.

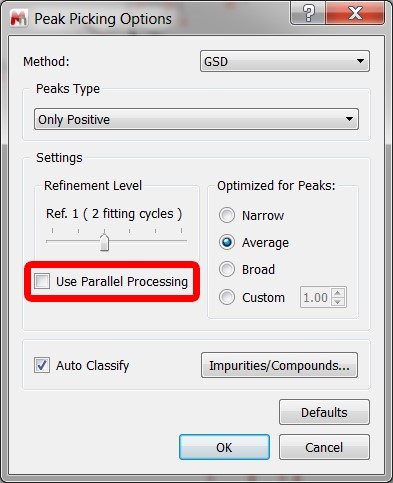
- GSD Parallelization.

- Capability to report the “Processing Template”.
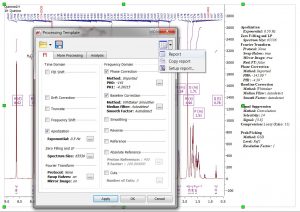
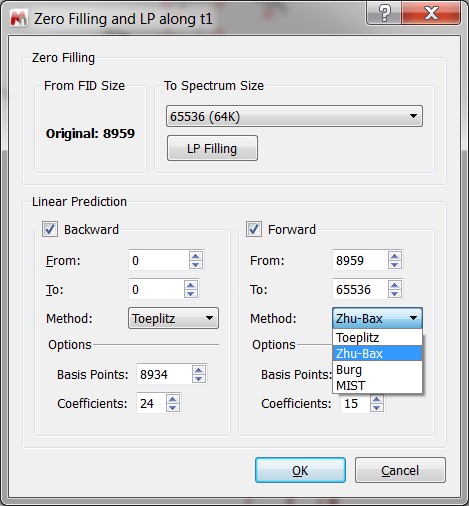
- Zero Filling and LP dialog has been improved.

- The Zero Filling part of the dialog indicates from which point the zero filling starts in cases of:
- Truncated FID
- The dimension f1 of phase-sensitive 2D spectra. - The forward and backward LP are no longer shared, because:
- The LP methods can be different.
- Not all LP methods are supported in backward LP.
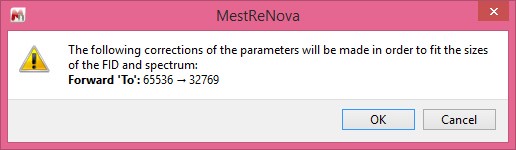
- Base points and number of coefficients can also be different. - If some LP parameters don’t match the sizes of the FID and spectrum then instead of auto-fitting them, a message box with the suggested corrections will appear as follows:

- Data Browser display experiment types.

- 1D traces in 2D spectra are updated automatically after applying Absolute Reference Script
- Import Directory Stack has been redesigned. A new version of the script with new features has been developed.
- Capability to select color of integral regions in Data Analysis.
- Auto track moving peaks in Data Analysis.
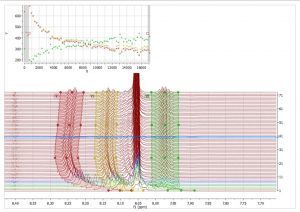
- Correction for gradient non-linearity in DOSY processing. Non-uniform gradient (NUG) correction can be applied by checking the option “Correct for non-uniform gradients” in Bayesian DOSY Transform dialog.

- Capability to open CNMR JCAMP-DX from Wiley.
- Fit traces to highest compound when added automatically by onTracesImported script.
- Ability to setup 1D spectra as a trace in superimposed 2D spectra via drag&drop.
- Import more parameters from Bruker TopSpin JCAMP files.
- Special processing for the NUTS Type 1 files produced with Oxford Instruments software.
- NUS processing has been improved so that 2D FID’s can now (optionally) be reconstructed during importing.
- Support NUS spectra in JEOL format.
- Drag and drop compatibility with new data storage format from VNMRJ3.2.
- FID reconstruction for Pure Shift experiments.
- 1D NMR autophase routine: New “Region analysis” algorithm. This new algorithm improves considerably the automatic phase routine for 1D NMR spectra. It can be selected on the Automatic Phase Correction options.
- Molecule Editor
-
- Show arrows for HMBC correlations with heteronuclei (15N, 29Si, etc.).
- Chemical structure in a pop-up window.
- Show PhysChem properties in columns.
- Ability to rotate fragments of a molecule.
- Ability to show correlation arrows by hovering the mouse over each atom.
- Prediction and Assignments data kept after editing the molecule with some of the commands (Normalize Numbering, Normalize Explicit Hydrogens Numbering, Concatenate Numbering, Add/Remove/Normalize Explicit Hydrogens)
- Change the background color when selecting a cell in Compounds accordionle.
- Ability to change the font size of atom numbers from the molecule in Compounds accordionle.
- Rotating function.
- Mirror function (top-down, left-right).
- Split a molecule and work with the parts individually.
- Ability to edit a molecule from the Compounds accordionle.
- Capability to draw unknown stereobonds.
- accordionle of Compounds opens automatically when importing a molecule.
- Read molecule parameters from JCAMP.
- Table of Compounds opens automatically when importing a molecule (optional)
- Ability to merge selected fragments
- Verify
-
- 2D peak picking independent of the visual threshold.
- DB
-
- Capability to add flags to daaccordionase field definitions: Now it is possible to set a flag as ‘Ediaccordionle’, ‘Required’ or ‘Unique’: Ediaccordionle: a field set as Ediaccordionle will be ediaccordionle after being stored in the DB. Required: a field set as Required will need to have a value when saved to a DB. Unique: a field set as Unique will be used to check for duplicate records when saving.

- Set field ‘Preview’ instead of ‘Chemical Structure’ from Molecule item to be shown in DB Browser.
- Port property in the DBClient.
- Need to wrap texts when displaying fields with long text string.
- Auto-scale of row height to the content of the field.
- Sample scripts for peak search and multiplet search in DB.
- Ability to store UV/DAD and ELSD chromatograms in a DB.
- Capability to add flags to daaccordionase field definitions: Now it is possible to set a flag as ‘Ediaccordionle’, ‘Required’ or ‘Unique’: Ediaccordionle: a field set as Ediaccordionle will be ediaccordionle after being stored in the DB. Required: a field set as Required will need to have a value when saved to a DB. Unique: a field set as Unique will be used to check for duplicate records when saving.
- Physchem
-
- PhysChem properties module: Explanatory, scientifically sound, statistically validated, quantitative structure-property prediction at your fingertips now integrated in Mnova (This work has been done in collaboration with Origenis GmbH). Consolidated models for chemists in the drug discovery space and beyond. We include “standard” properties such us:
- logP
- Molecular Volume
- Contact Surface Polar HBD
- Contact Surface Polar HBA
- Contact Surface Neutral
- Contact Surface Total
- logS
- Abraham A
- Abraham B
- Abraham E
- Abraham S
- Abraham V
- Boiling Point
- Henry’s Law Constant
- Hydroxy Radical Attack
- Melting Point
- Vapor Pressure
- logD
In addition we provide properties impossible to find in any other commercial package:
- Human Serum Albumin Affinity Constant
- Alpha Acid Glycoprotein Affinity Constant
- Membrane Affinity Constant
- Per cent Plasma Protein Binding
- Free Plasma Fraction
- Blood-Brain-Partition Coefficient
- Free Brain Fraction
In addition, if you have data of a particular property you are interested in, we can build a model for you.
- PhysChem properties module: Explanatory, scientifically sound, statistically validated, quantitative structure-property prediction at your fingertips now integrated in Mnova (This work has been done in collaboration with Origenis GmbH). Consolidated models for chemists in the drug discovery space and beyond. We include “standard” properties such us:
- Scripting Engine
- There are many improvements in our Scripting Engine tools. You can see below a summary of some of main ones among many more. Please do contact us if you are interested in any particular area related to your analytical applications and we will give you a more comprehensive and personalized piece of information:
- Pixmap feature. The new Pixmap object is an off-screen image representation that can be used as a paint device.
- Capability to write odf files. The new Text Document object makes possible to create OpenDocument Format (ODF) files from any text.
- Added zip/unzip features. The new Zip and Unzip objects can be used to create and manage compressed files.
- Added Printer object. The Printer object is a paint device that paints on a printer.
- New network features. This object NetworkAccessManager allows the application to send network requests and receive replies.










Other advanced features
- Denoising algorithms for 1D and 2D NMR spectra.
- Improved manual multipoint baseline correction method.
- Enhanced Group Delay correction algorithm for Bruker spectra.
- Improved GSD algorithm.

{kind=link}
{kind=link}