
Accurate prediction of 1H and 13C NMR spectra from a chemical structure.
Mnova NMR Predict calculates accurate and precise NMR chemical shifts using a novel procedure that combines several prediction engines in a constructive way. This method is called Ensemble NMR Prediction and uses several Machine Learning methods in combination with the well-knonwn Increments and HOSE-code algorithms developed by Modgraph Consultants. Prediction of chemical shifts of other nuclides is also available.
To complement the article about Ensemble NMR Prediction you can also read a blog post about 1H data here
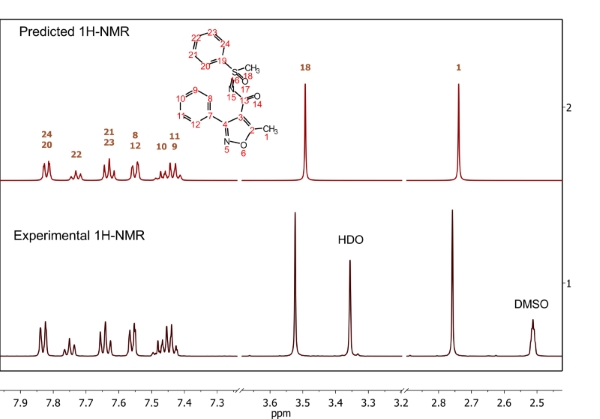

Easily combine and compare experimental and predicted data as part of your workflow.
Mnova NMR Predict: 45-day FREE trial
 1. Download
1. Download
A plugin integrated in Mnova (separate license). No extra installer is required.
 2. Installation
2. Installation
Open Mnova and go to ‘Help/Get-Install Licenses’. Select ‘Evaluate’.
 3. License
3. License
Fill in the form to receive your trial license via e-mail.
Help & Resources
 Videos
Videos
Please note that the new Mnova NMRPredict “Ensemble NMR Predictor” (Version ≥ 14) consists in two different licenses, Modgraph and Mestrelab Predictors
NMRPredict
Highlights

Make better decisions for your spectra faster!


Academic, Government & Industrial
Markets
- Pharmaceutical, chemical and food industries and QC environments
- Research and NMR teaching in Academia
- Suitable for individual users, research groups as well as large institutions and companies







